Frontiers |
您所在的位置:网站首页 › tunica intima › Frontiers |
Frontiers
MINI REVIEW article
Front. Immunol., 12 September 2022Sec. Molecular Innate Immunity
Volume 13 - 2022 |
https://doi.org/10.3389/fimmu.2022.976722
Vascular homeostasis in atherosclerosis: A holistic overview Suowen Xu1*†‡, Suowen Xu1*†‡,  Qing Rex Lyu2†‡, Qing Rex Lyu2†‡,  Iqra Ilyas1, Iqra Ilyas1,  Xiao-Yu Tian3†‡ and Jianping Weng1**‡1Department of Endocrinology, Institute of Endocrine and Metabolic Diseases, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, Clinical Research Hospital of Chinese Academy of Sciences (Hefei), University of Science and Technology of China (USTC), Hefei, China2Medical Research Institute, Chongqing General Hospital, Chongqing, China3School of Biomedical Sciences, Chinese University of Hong Kong, NT, Hong Kong SAR, China Xiao-Yu Tian3†‡ and Jianping Weng1**‡1Department of Endocrinology, Institute of Endocrine and Metabolic Diseases, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, Clinical Research Hospital of Chinese Academy of Sciences (Hefei), University of Science and Technology of China (USTC), Hefei, China2Medical Research Institute, Chongqing General Hospital, Chongqing, China3School of Biomedical Sciences, Chinese University of Hong Kong, NT, Hong Kong SAR, ChinaAtherosclerosis refers to the deposition of lipids and the co-existence of inflammation and impaired inflammation resolution in pan-vasculature, which causes lumen narrowing, hardening, plaque formation, and the manifestation of acute cardiovascular events. Emerging evidence has suggested that vascular circulation can be viewed as a complex homeostatic system analogous to a mini-ecosystem which consists of the vascular microenvironment (niche) and the crosstalk among phenotypically and functionally diverse vascular cell types. Here, we elucidate how cell components in the vascular wall affect vascular homeostasis, structure, function, and atherosclerosis in a holistic perspective. Finally, we discuss the potential role of vascular-stabilizing strategies including pharmacotherapies, natural substances and lifestyle modifications, in preventing cardiovascular diseases by preserving vascular integrity and homeostasis. Atherosclerosis and cardiovascular risk factorsAtherosclerosis is a complex and progressive disease with the interplay of multiple cell types and mechanisms involving genetic, epigenetic, environmental, metabolic, clonal hematopoiesis, and lifestyle factors and an evolving landscape (1) (Figure 1A). Pan-vascular atherosclerosis is the major cause of cardiovascular disease (CVD) and ischemic stroke (1–3). Atherosclerosis preferentially develops in medium- and large-sized arteries. The blood vessel wall normally consists of tunica intima, tunica media, tunica adventitia and tunica adiposa (4). The blood vessel microenvironment resembles a mini-ecosystem which consists of different cell types, including cells in the blood (red blood cells, leukocytes, monocytes etc.) and vascular cells [endothelial cells (ECs), vascular smooth muscle cells (VSMCs), macrophages and T-lymphocytes etc.)]. These cell types crosstalk with each other via ligand-receptor interaction, exosome, endothelial cell secretome as well as cell-matrix interactions, thereby regulating vascular tone and stabilize tissue homeostasis (Figure 1B). FIGURE 1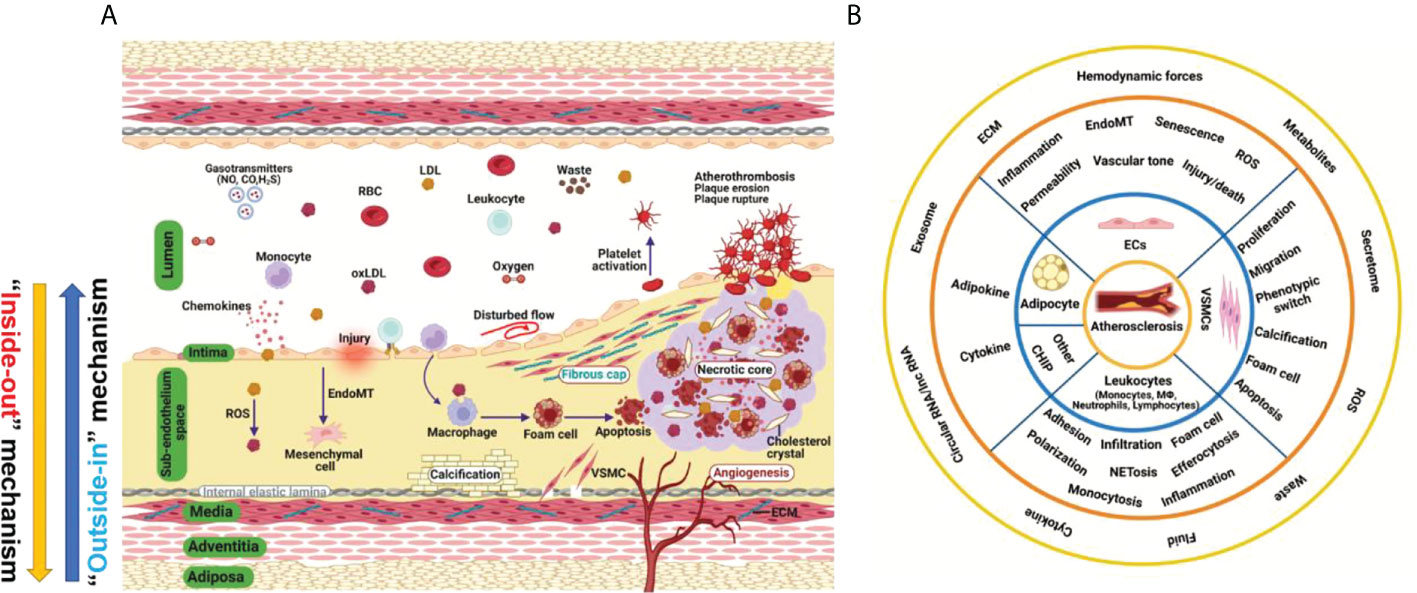 Figure 1 Hallmark of atherosclerosis. (A) The pathogenesis of atherosclerosis. Vascular components of atherosclerotic artery which include tunica adiposa, tunica adventitia, tunica media, sub-endothelium space and tunica intima. For simplicity, external elastic lamina in the blood vessel was not depicted. The interplay of different vascular cell types within the local vascular niche drives atherosclerosis development. Both “inside-out” and “outside-in” mechanism contribute to the development of atherosclerosis. (B) A holistic overview of hallmarks of atherosclerosis and crosstalk among vascular cell components. Atherosclerosis is complex disease involving different cell types at different vascular layers. The hallmark events in different cell types act in concert to promote atheroprogression. CHIP, clonal hematopoiesis of indeterminate potential; ECs, endothelial cells; ECM, extracellular matrix; EndoMT, endothelial-mesenchymal transition; ROS, reactive oxygen species; VSMC, vascular smooth muscle cells. Recognized traditional and non-traditional risk factors for CVD include: hyperlipidemia, hypertension, hyperglycemia, hyperuricemia, infection, smoking, male gender, high triglyceride-rich lipoproteins, lipoprotein (a), sleep deprivation, physical inactivity, gut microbiome imbalance, environmental pollution, noise, and psychological stress (1). Taking exercise for example, a recent study (5) has demonstrated that moderate exercise promotes cardiovascular health; however, long-term strenuous exercise damages tunica media, increases aortic stiffness, causes elastic lamina to rupture and media layer thickening of intramyocardial arteries. The authors also found that deleterious remodeling rendered by intensive exercise persisted after withdrawal from exercise (5). Advanced understanding of the novel mechanistic basis of atherosclerosis by addressing how these risk factors causes atherosclerosis will open new avenues to therapeutic interventions aiming to prevent and treat atherosclerosis. In the following section, we summarize the role of cell components in the vascular wall in vascular microenvironment homeostasis and atherosclerosis, with an aim to providing a unified and holistic overview of future research directions in this area. Vascular wall components in vascular homeostasis and atherosclerosisEndothelial cells in the tunica intima as sentinels in vascular health and pivotal regulator of atherosclerosisECs are the key cell type that connects circulating blood and the vascular wall. ECs serve as a natural barrier in the pan-vascular system (6). ECs sense the mechanosignals via mechanosensing complex or mechanosensors like plexin D1 (7). ECs serve as the sentinel as well as safeguard for vascular health and tissue homeostasis. The vascular endothelium is thus deemed as a protype of homeostatic regulation of endothelial function (8). Following insults, ECs initiate powerful backup systems/mechanisms to prevent vascular injury. As long as vascular injuries are not overwhelming and reparable, existing cellular defense machineries will be working to maintain vascular homeostasis (8). However, when the injury persists and ECs become activated, leading to endothelial dysfunction, such as inflammation, hyperpermeability, leukocyte adhesion, and cell senescence (6, 9). The earlier stage of low-degree of EC activation is a compensatory tissue repair mechanism that promotes endothelial rehabilitation and preserves vascular health. However, persistent activation of ECs prepares ECs to transit into a decompensatory stage, which leads to endothelial dysfunction (6). After the initial stage of endothelial dysfunction, ECs will recruit more leukocytes to the site of injury, followed by cell adhesion, rolling, crawling, and diapedesis that contributes to atherosclerosis development (6). It is proposed that the protective functions rendered by ECs at resting states include the maintenance of EC quiescence, anti-inflammation, anti-oxidation and anti-proliferation (10). Beyond canonical physiological functions, ECs also emerge as a new type of innate immune cells (11) which execute a plethora of immune functions, such as cytokine production, phagocytosis of cell debris and bacteria, sensing danger and pathogen-associated molecular patterns, antigen presentation, immune metabolism and trained immunity (11, 12). For example, trained immunity is an immunological concept which refers to long-lasting pro-inflammatory phenotype (similar as innate immune memory) after prior stimulation with or exposure to microbial products and pro-atherogenic stimuli (such as β-glucan, aldosterone, oxidized LDL and LPS), leading to sustained hyperactivation of innate immune system (13). Trained immunity is an important biological mechanism which contribute to atherosclerotic cardiovascular disease via epigenetic modification and reprogramming immune-metabolism (13, 14). Recent studies have revealed that, upon infection with virus and bacteria, the anti-viral signaling pathway is activated, the RIG-I (retinoic acid-inducible gene I) pathway in particular (12). Infection with coronavirus, such as SARS-CoV-2, can cause trained immunity in ECs, potentially leading to enhance inflammation in ECs (15). ECs in their native state also maintain anti-thrombotic and hemostatic functions, which prevent platelet activation and adhesion to activated endothelium. In addition, ECs have active secretory pathways, vesicles and granules to produce huge proteomes to maintain vascular homeostasis and integrity (16). Secreted proteins from ECs play an important role in regulating vascular ECs crosstalk with other vascular cell types. The local microenvironment (niche) in vascular wall and cardiovascular disease conditions can modify endothelial function by regulating ECs’ secretome. Recently, quantitative proteomic analysis of the endothelial secretome (17) has greatly accelerated the discovery of novel secreted proteins from ECs which regulate vascular homeostasis and atherosclerosis. In addition, EC secretome also provides promising biomarkers which have diagnostic and prognostic value for various vascular disorders (16). More importantly, ECs are the innermost layer of cells that are directly exposed to a wide variety of cardiovascular risk factors, including hyperglycemia, hyperlipidemia, hyperuricemia, hyperhomocysteinemia, and cigarette smoking (12). In addition, a recent study has demonstrated an intimate relationship between cholesterol homeostasis and inflammation/immunity in ECs by showing that pro-inflammatory cytokines upregulated SREBP2 (sterol regulatory element binding protein 2) cleavage/activation and augmented the expression of genes involved in cholesterol biosynthesis via NF-kB (nuclear factor-kappa B) and SREBP2 pathway (18). Other biological functions of ECs include the secretion of nitric oxide (NO) and other vasoactive molecules, such as hydrogen sulfide (H2S), prostacyclin (PGI2), endothelin-1 (ET-1) and endothelium-derived hyperpolarization factor (EDHF) (6). By balanced production of these molecules, ECs regulate vascular tone. Furthermore, ECs are highly heterogenous and plastic cell types that can acquire a mesenchymal-like phenotype through endothelial–mesenchymal transition (EndoMT) process via the TGFβ/Smads, YAP/TAZ, Snail, Twist, and ZEB family of transcription factors. Undergoing EndoMT, EC-specific markers are lost, and mesenchymal cell-specific markers are acquired, thus impairing normal EC function. EndoMT is strongly implicated in the development of atherosclerotic plaques in mice and in huma patients (6). Endothelial homeostasis is regulated by several master transcription factors, such as KLF2, KLF4 and Nrf2, the activation of which leads to anti-inflammatory, anti-oxidant, anti-thrombotic and inflammation-resolution effects (10, 19). Also, upon injurious stimuli exposure (such as aging, irradiation, anti-cancer therapy, inflammation, virus/bacterial infection and cigarette smoking etc.), ECs will become senescent, and senescence-associated secretory phenotype (SASP) will be acquired, leading to inflammation and dysfunction of neighboring cells (6). The concerted actions of multiple cardiovascular risk factors will cause endothelial cell death and cell loss, leading to plaque erosion and subsequent formation of plaque rupture. Altogether, ECs function far beyond a physical barrier and participate in vital processes in atherosclerosis (20). Sub-endothelium space: The fertile “soil” of atherosclerosisAtherosclerosis preferentially develops in the subendothelial space (SES) of large- and medium-sized arteries in aortic regions with oscillatory blood flow. The SES is a fertile “soil” (microenvironment or niche) for atherosclerosis development due to the intricate interplay among subendothelial low-density lipoprotein (LDL) retention, LDL transcytosis, VSMC proliferation and migration, endothelial dysfunction, foam cell formation, and necrotic core formation (21). After endothelial injury, VSMCs migrate from the media layer to SES and secrete pro-fibrotic extracellular cellular matrix proteins, such as collagens, and fibronectins, leading to vascular remodeling and intimal thickening (21). LDL can pass injured endothelium via passive transport or receptor-mediated transcytosis across vascular endothelium. Afterward, LDL can be retained in the SES or oxidatively modified to be oxLDL, which was subsequently uptaken by VSMCs or macrophages to form foam cells, the hallmark of atherosclerosis (22). A proliferation-inducing ligand (APRIL) is a cytokine that binds to heparan-sulfate proteoglycans (HSPGs). APRIL exerts atheroprotective effects by binding to heparan sulfate chains of HSPG2, thus limiting LDL-C retention in the vessel wall, reducing macrophages content and size of necrotic cores (23). Infiltrated monocytes also differentiate into macrophages, which is a classical innate immune cell type that mediates inflammatory response in SES. A recent study (24) has shown that the olfactory receptor Olfr2 (human ortholog olfactory receptor 6A2, OR6A2) can detect octanal in the circulating blood, leading to interleukin-1β (IL-1β) in macrophages by activating NLRP3 inflammasome. In addition to mediating the activation of inflammasome and inflammation (25), macrophages can also become senescent, and senescent macrophages can accumulate in SES and drive the expression of pro-atherogenic and pro-inflammatory cytokines/chemokines, thus favoring features of plaque vulnerability, including the thinning of fibrous cap and fragmentation of elastic fibers. In contrast, selective clearance of these senescent cells by senolytics prevents atheroma formation (26). Other mechanisms include lipid toxicity induced by free cholesterol and cholesterol crystal, continued buildup of lipid, impaired inflammation resolution, and infiltration of immune cells such as CD3+ and CD4+ T lymphocytes, fan the flame of inflammation, cell death, and atherosclerosis (15), leading to plaque necrosis and vulnerability. Within the plaques, macrophages are the main decomposing machinery in the atherosclerotic plaque that clears necrotic cell debris via the efferocytosis mechanism (27). Other decomposing machinery include autophagy as well as chaperone-mediated autophagy (CMA). Recent evidence has suggested that defective CMA via LAMP2A (lysosome-associated membrane protein type 2A) occurs in mouse and human vasculature and that decline of CMA in VSMCs and macrophages promote NLRP3 inflammasome activation, metabolic dysfunction and atherogenesis (28–30). VSMCs in tunica media as a key mediator of atherosclerosisThe tunica media consist of multiple layers of VSMCs and is interval-arranged with the elastin lamina. VSMCs are the main scaffold cell type that constitutes tunica media, which is essential for the optimal functioning of arteries by eliciting vasodilation (31). The proliferation and migration of VSMCs are strongly impacted by interaction of VSMCs with extracellular matrix (ECM) proteins, such as proteoglycans, fibronectin, collagen and elastic fibers (32). ECM dysregulation is an important mechanism for disrupted vascular function, atherosclerosis and aortic dissection (32). VSMCs are highly plastic with at least two phenotypes, contractile and synthetic VSMCs, which play an essential role in the embryonic development of vasculature and onset of various diseases (31). Under basal conditions, VSMCs are in the quiescent stage, which is less proliferative and has a relatively low turnover rate. Upon vascular injury, such as chronic hypercholesterolemia or endothelial denudation by drug-eluting stents, the contractile VSMCs switch to synthetic phenotype and undergo proliferation, as well as migration from vascular media to the injury site, to propagate wound repairing (31). During this process, the synthetic VSMCs secrete abundant cytokines and extracellular matrix proteins to create a favorable microenvironment for cell migration and vascular remodeling. During this phenotypic switch, the expression of multiple VSMCs-specific markers is decreased, such as α-smooth muscle actin (SMA), smooth muscle 22 α (SM22α), calponin 1 (CNN1), smooth muscle myosin heavy chain (SM-MHC) and transform to the synthetic phenotype, which contributes to neointima hyperplasia, atherosclerosis and aortic aneurysm (31). In addition, the expression of contractile marker SM22α blocks VSMC-derived foam cell formation via augmenting liver X receptor (LXR)-dependent cholesterol efflux (33). The expression of genes associated with the contractile phenotype of VSMCs is regulated by multiple transcription factors, epigenetic regulators, and noncoding RNAs, such as Myocardin/SRF (34), TET2 (35), SENCR (36), MYOSLID (37), and CARMN (38). While the synthetic phenotype of VSMCs is transcriptionally and epigenetically regulated by krüppel-like factor 4 (KLF4) (35) and NEAT1 (39). In addition to this phenotypic switch, VSMCs can also undergo calcification by augmented expression of calcification-related proteins, such as osteopontin (OPN, also known as SPP1), bone morphogenetic proteins (BMPs), alkaline phosphatase (ALP), the expression of which are regulated by ERK1/2, Runt-related transcription factor 2 (RUNX2) and Wnt signaling pathway (20). Besides, VSMCs can also form foam-like cells after loading with cholesterol, and this source of foam cells from VSMCs has long been underestimated for their role in atherosclerosis (40). Specifically, by genetic inducible fate mapping in mice, medial VSMCs can lose classical VSMC marker genes and transdifferentiate into macrophage-like cells and mesenchymal stem cells in a KLF4 -dependent manner, suggesting high VSMC plasticity in the development of atherosclerosis (41, 42). Single-cell RNA-sequencing studies have confirmed that VSMCs are highly heterogenous, possessing different phenotypes, including senescent, foam cell-like, and osteoblast-like phenotypes (20). Future studies should be directed to elucidate the interaction between VSMCs and vascular niche and how this interaction instructs atherogenesis (20). The tunica media could be one of the most critical layers in atherosclerosis development. On one hand, being localized underneath the tunica intima, VSMCs are shielded from direct contact with the bloodstream unless the endothelial monolayer is compromised (43). The integrity of the tunica intima fundamentally influences the behavior of VSMCs. Once injured, monocytes could infiltrate into the tunica media and transform into macrophages, which release multiple cytokines and/or growth factors that re-shape the VSMC phenotype (43). Moreover, the platelets could be activated upon vascular injury and release a large number of inflammatory cytokines and chemokines, which induces the phenotypic switch of VSMC from contractile to synthetic phenotype (31). On the other hand, interventions in adventitia also significantly impact tunica media. For example, direct elastase immersion on the adventitia of the aorta could eventually result in the formation of the aortic aneurysm (44). Therefore, it is important to protect tunica media from being disrupted by the external factors (including dietary, environmental factors) from either inner or outer layers of the blood vessel. It also suggests that the progression of atherosclerosis, could be very difficult to alleviate unless VSMC-derived factors are properly controlled. Tunica adventitia and tunica adiposa: The “wonderland” of atherosclerosisThe tunica adventitia consists of a more complex mixture of multiple cell types, including fibroblasts, pericytes, macrophages, T cells, dendritic cells, and mast cells etc (4). Most atherosclerosis research has been focused on studying the role of ECs (intimal cells), macrophages/immune cells (SES in disease vessel), VSMCs (media layer cells) in atherosclerosis (20); however, the role of adventitial components and derived factors in atherosclerosis has long been understudied. It is assumed that the cells in the adventitia offer supporting functions to the blood vessel and can also regulate the structure and function of other vascular cells by cell-cell communications, exosome, secretome (such as growth factors, angiocrine factors, cytokines/chemokines, and vasoactive peptides/hormones) etc. (20). Adventitial cells are involved in regulating vascular tone and blood pressure as well as atherosclerosis via paracrine and endocrine signaling (45). As plaques in the SES are devoid of innervation. A recent study has elegantly shown that the peripheral nervous system utilizes the adventitia as the main conduit to directly interact with diseased arteries to regulate atherosclerosis via the existence of neuroimmune cardiovascular interfaces (NICIs) in adventitia segments (46). Also, another intriguing and largely unexplored research question is the precise role of perivascular adipose tissue (PVAT) in vascular health and diseases, such as atherosclerosis (45, 47). Recent evidence has implicated PVAT as the fourth layer of the blood vessel wall (the tunica adiposa), which produces adipokines and vasoactive substances (such as leptin, adiponectin, resistin, and visfatin)/cytokines/growth factors that regulate vascular tone/homeostasis (4, 48). One study from Tang laboratory have revealed that periaortic knockdown of ribosomal protein S3A (RPS3A) in mouse PVAT impaired PVAT browning, promoted vascular inflammation and atherosclerosis development by modulating UCP-1 expression in ApoE-/- mice (49). Similarly, another recent study from the Tang laboratory demonstrated that BMP4 (bone morphogenetic protein 4) depletion in PVAT increased endothelial inflammation in an EC/adipocyte co-culture system via amplifying IL-1β-driven inflammatory response. More importantly, BMP4 deficiency in adipose tissues exacerbates atherosclerosis, while BMP4 overexpression in adipose tissues promotes PVAT browning and atheroprevention in ApoE-/- mice (50). These findings uncover the important role of PVAT in regulating endothelial function, inflammation and atherosclerosis. Future studies including PVAT in measuring aortic wall’s mechanical behavior, such as aortic stiffness and vasorelaxation/constriction is important (48). The emergence of single-cell RNA-sequencing and the generation of PVAT-specific Cre to precisely manipulate PVAT-derived factors will be useful research tools to answer this question due to the fact that tunica adiposa is a mixture of heterogenous adipocytes (including white, brown and beige adipocytes) (4). The endocrine and paracrine functions of PVAT-derived factors in “outside-in” mechanism of atherosclerosis is an important research direction (4). Strategies that stabilize the vascular system to prevent atherosclerosisBased on the pivotal role of deregulated vascular homeostasis in atherosclerosis, the “multiple hits” hypothesis of atherosclerosis is gaining more evidence (Figure 2A). To this end, several categories of drugs are able to stabilize the “vascular niche” (51) and prevent atherosclerosis (Figure 2B). These drugs include statins (lipid-lowering drugs) (52), Angptl3 inhibitors (53), ACLY inhibitors (such as Bempedoic acid) (54), gliflozins (SGLT2 inhibitors, anti-diabetic drugs) (55), glutides (GLP-1 receptor agonists, anti-diabetic drugs) (56), metformin (anti-diabetic drugs) (57–59), aspirin (COX inhibitor, NSAIDs), Angiotensin II converting enzyme inhibitors (ACEI, anti-hypertensive drugs) (60), Angiotensin II receptor blockers (ARBs, anti-hypertensive drugs) (60, 61), naturally-occurring NLRP3 inflammasome inhibitor (colchicine) (62), KLF2 activators (63, 64), AMPK activators (endothelial protective drugs) (65) and many others. Based on the complex nature of atherosclerosis, polypill or ploypharmacology targeting established mechanisms/risk factors are needed. Vasoprotective effects of these drugs include maintenance of vascular homeostasis by generating NO, maintaining normal vascular tone, reducing oxidative stress, inhibiting NF-κB-dependent pro-inflammatory response, and reducing the production of vasoconstrictive molecules (such as AngII and ET-1) as well as stabilizing glycocalyx microstructure in vascular endothelium (8). These strategies also reduce foam cell formation in macrophages by limiting lipid uptake and augmenting cholesterol efflux, attenuating macrophage inflammation and skewing from M1 pro-inflammatory phenotype toward tissue-repairing M2 phenotype (66). In addition, these strategies also halt VSMC proliferation, migration and phenotypic switch from contractile to synthetic phenotype (43). Detailed studies in assessing the effects and mechanism of action of these pharmacotherapies in cell-cell crosstalk warrants further studies. FIGURE 2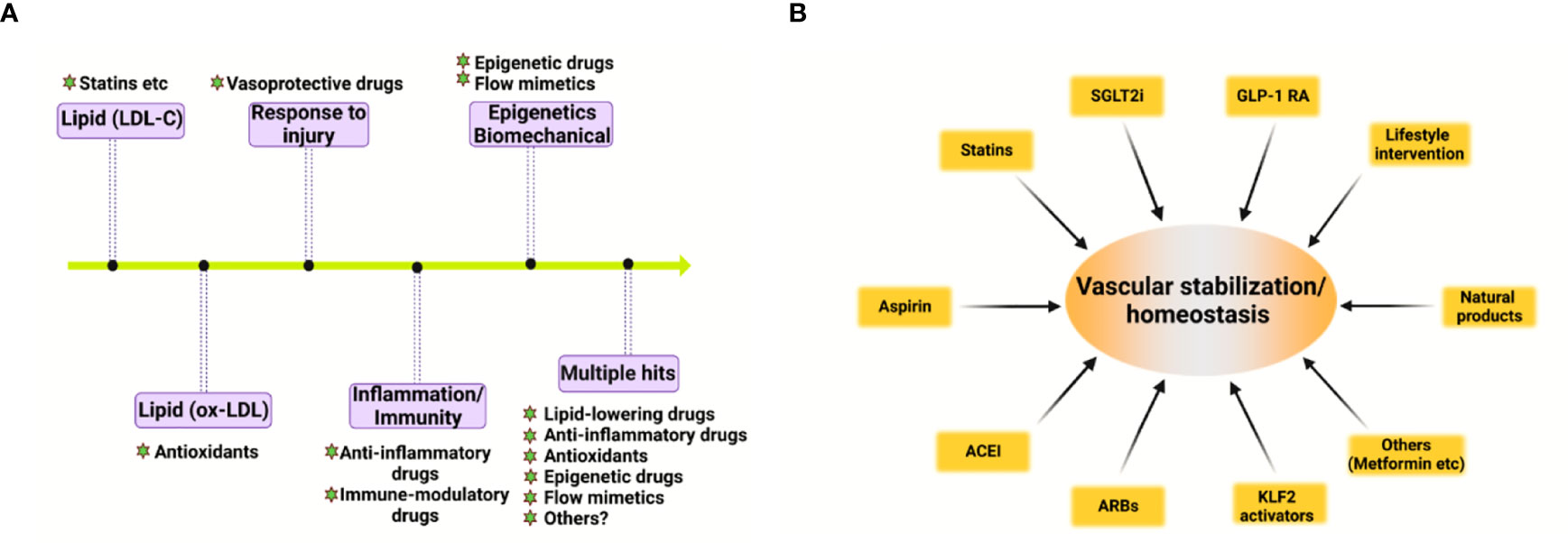 Figure 2 Evolving hypothesis of atherosclerosis and vascular homeostasis-targeted therapies. (A) Evolving hypothesis of atherosclerosis. (B) Pharmacological and non-pharmacological strategies to promote vascular stabilization and homeostasis and to prevent atherosclerosis. ACEI, angiotensin-converting-enzyme inhibitors; ARB, angiotensin II receptor blockers; SGLT2i, sodium-glucose cotransporter 2 inhibitor; GLP-1 RA, glucagon-like peptide 1 receptor agonist; KLF2, kruppel-like factor 2. In addition to these drugs, lifestyle modifications, such as exercise, healthy habitual eating, smoking cessation also maintains vascular homeostasis and prevent atherosclerotic cardiovascular diseases (67, 68). Therefore, pharmacological and non-pharmacological strategies aimed at recuperating vascular stabilization and homeostasis hold promise for anti-atherosclerotic therapies (8). Concluding remarks and future perspectivesAtherosclerosis is a progressive disease with a changing landscape which needs to be understood in a holistic view (1). The main theories of atherosclerosis have changed considerably during the past decades. It is increasingly recognized that lipid (both LDL-cholesterol and triglyceride-rich lipoproteins) and inflammation are two predominant mechanisms of atherosclerosis (1). However, the development of atherosclerosis is a slow but progressive process with the interplay of multiple mechanisms. Therefore, a “multiple-hit” hypothesis is possible, including mechanisms of an aberrant lipid profile, inflammation, vascular injury, oxidative stress, hemodynamic forces, epigenetics and others (Figure 2A). In this hypothesis, different vascular cell types interact with each other via ligand-receptor pairs or proteins in the secretome or cargos (such as miRNAs, circular RNAs, and lncRNAs) carried by exosomes (Figures 1A, B). The “multiple-hit” hypothesis of atherosclerosis expands previous established “response-to-injury”, “inflammation”, “LDL oxidation” hypothesis (1), and involve both the “inside-out” (intima-subendothelium-media-adventitia-adiposa) and “outside-in” (adiposa-adventitia-media-subendothelium-intima) mechanisms (Figure 1A). This bidirectional mechanism of atherosclerosis reminds us the need to consider cell-cell/cell-environment interaction in atherosclerosis research. In this article, we reviewed the biological functions of the structural components of the vessel wall in the vascular homeostasis and atherosclerosis. Considering that atherosclerosis is a focal disease that preferentially develops in regions where disturbed blood flow occurs, and the flow pattern is turbulent/oscillatory flow (10, 19), the spatio-temporal and biomechanical basis of atherosclerosis remain to be validated (69). The focal nature of atherogenesis resembles the zonation phenomenon in the liver, which is coordinated by multiple cell types (70, 71). We term this phenomenon “aorta zonation”. Tentatively, the aorta can be categorized into 5 zones, zone 1 is the inner curvature of the aortic arch; zone 2 is the greater curvature of the aortic arch; zone 3 is the arterial branches of the aortic arch; zone 4 is the thoracic aorta; zone 5 is the rest of arterial branches in the aorta. Zone 1, 3, and 5 are predilection sites of atherosclerosis development; however, zone 2 and 4 are protected against atherosclerosis. It is speculated that vascular cells in zone 1 to zone 5 are highly heterogenous and plastic, with different genetic, epigenetic, and metabolic traits. Systems biology techniques such as next-generation single-cell spatial transcriptomics will be an important tool to delineate zone-specific gene expression programs in the aorta and further understand the mechanistic basis of the heterogeneity of vascular cells in different aortic zones and reveal novel therapeutic targets for atherosclerosis. Last but not least, it is of vital importance to elucidate how the vascular microenvironment/niche instructs vascular health and diseases and how homeostatic control of vascular function is achieved. It is well established that atherosclerosis arises and progresses when the homeostatic control is perturbed-pro-atherogenic signaling overwhelms anti-atherosclerotic signaling, leading to complex regulatory loops signaling and atheroprogression (69). Basic and translational research into the mechanisms of maintaining vascular homeostasis raises the exciting prospect of alleviating the ever-growing burden of atherosclerosis by addressing residual cholesterol and inflammation risk in patients with CVD. Author contributionsSX and JW conceptualized the review. SX and QL wrote the review. II made figures and edits the manuscript. X-YT provides intellectual input and edits on the manuscript. All authors contributed to the article and approved the submitted version. FundingThis study was supported by grants from National Key R&D Program of China (No. 2021YFC2500500), National Natural Science Foundation of China (Grant Nos. 82070464, 81941022, 81530025) and Strategic Priority Research Program of Chinese Academy of Sciences (Grant No. XDB38010100). This work was also supported by Program for Innovative Research Team of The First Affiliated Hospital of USTC (CXGG02), Anhui Provincial Key Research and Development Program (Grant No. 202104j07020051) and Anhui Province Science Fund for Distinguished Young Scholars (Grant No. 2208085J08). Figures were drawn with Biorender.com. Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher. References1. Libby P. The changing landscape of atherosclerosis. Nature (2021) 592(7855):524–33. doi: 10.1038/s41586-021-03392-8 PubMed Abstract | CrossRef Full Text | Google Scholar 2. Björkegren JLM, Lusis AJ. Atherosclerosis: Recent developments. Cell (2022) 185(10):1630–45. doi: 10.1016/j.cell.2022.04.004 PubMed Abstract | CrossRef Full Text | Google Scholar 3. Zhou X, Yu L, Zhao Y, Ge J. Panvascular medicine: an emerging discipline focusing on atherosclerotic diseases. Eur Heart J (2022). doi: 10.1093/eurheartj/ehac448 CrossRef Full Text | Google Scholar 4. Hillock-Watling C, Gotlieb AI. The pathobiology of perivascular adipose tissue (PVAT), the fourth layer of the blood vessel wall. Cardiovasc Pathol (2022) p:107459. doi: 10.1016/j.carpath.2022.107459 CrossRef Full Text | Google Scholar 5. Rubies C, Batlle M, Sanz-de la Garza M, Dantas AP, Jorba I, Fernandez G, et al. Long-term strenuous exercise promotes vascular injury by selectively damaging the tunica media: Experimental evidence. JACC Basic Transl Sci (2022) 7(7):681–93. doi: 10.1016/j.jacbts.2022.02.017 PubMed Abstract | CrossRef Full Text | Google Scholar 6. Xu S, Ilyas I, Little PJ, Li H, Kamato D, Zheng X, et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: From mechanism to pharmacotherapies. Pharmacol Rev (2021) 73(3):924–67. doi: 10.1124/pharmrev.120.000096 PubMed Abstract | CrossRef Full Text | Google Scholar 7. Mehta V, Pang KL, Rozbesky D, Nather K, Keen A, Lachowski D, et al. The guidance receptor plexin D1 is a mechanosensor in endothelial cells. Nature (2020) 578(7794):290–5. doi: 10.1038/s41586-020-1979-4 PubMed Abstract | CrossRef Full Text | Google Scholar 8. Huwiler A, Pfeilschifter J. Recuperation of vascular homeostasis. Circ Res (2021) 129(2):237–9. doi: 10.1161/CIRCRESAHA.121.319558 PubMed Abstract | CrossRef Full Text | Google Scholar 9. Wang L, Cheng CK, Yi M, Lui KO, Huang Y. Targeting endothelial dysfunction and inflammation. J Mol Cell Cardiol (2022) 168:58–67. doi: 10.1016/j.yjmcc.2022.04.011 PubMed Abstract | CrossRef Full Text | Google Scholar 10. Niu N, Xu S, Xu Y, Little PJ, Jin ZG. Targeting mechanosensitive transcription factors in atherosclerosis. Trends Pharmacol Sci (2019) 40(4):253–66. doi: 10.1016/j.tips.2019.02.004 PubMed Abstract | CrossRef Full Text | Google Scholar 11. Shao Y, Saredy J, Yang WY, Sun Y, Lu Y, Saaoud F, et al. Vascular endothelial cells and innate immunity. Arterioscler Thromb Vasc Biol (2020) 40(6):e138–52. doi: 10.1161/ATVBAHA.120.314330 PubMed Abstract | CrossRef Full Text | Google Scholar 12. Xu S, Jin T, Weng J. Endothelial cells as a key cell type for innate immunity: A focused review on RIG-I signaling pathway. Front Immunol (2022) 13:951614. doi: 10.3389/fimmu.2022.951614 PubMed Abstract | CrossRef Full Text | Google Scholar 13. Flores-Gomez D, Bekkering S, Netea MG, Riksen NP. Trained immunity in atherosclerotic cardiovascular disease. Arterioscler Thromb Vasc Biol (2021) 41(1):62–9. doi: 10.1161/ATVBAHA.120.314216 PubMed Abstract | CrossRef Full Text | Google Scholar 14. Zhong C, Yang X, Feng Y, Yu J. Trained immunity: An underlying driver of inflammatory atherosclerosis. Front Immunol (2020) 11:284. doi: 10.3389/fimmu.2020.00284 PubMed Abstract | CrossRef Full Text | Google Scholar 15. Shao Y, Saredy J, Xu K, Sun Y, Saaoud F, Drummer C.t., et al. Endothelial immunity trained by coronavirus infections, DAMP stimulations and regulated by anti-oxidant NRF2 may contribute to inflammations, myelopoiesis, COVID-19 cytokine storms and thromboembolism. Front Immunol (2021) 12:653110. doi: 10.3389/fimmu.2021.653110 PubMed Abstract | CrossRef Full Text | Google Scholar 16. Rusu L. Endothelial secretome. IntechOpen (2022). doi: 10.5772/intechopen.106550 CrossRef Full Text | Google Scholar 17. Zhao Y, Fang R, Zhang J, Zhang Y, Bechelli J, Smalley C, et al. Quantitative proteomics of the endothelial secretome identifies RC0497 as diagnostic of acute rickettsial spotted fever infections. Am J Pathol (2020) 190(2):306–22. doi: 10.1016/j.ajpath.2019.10.007 PubMed Abstract | CrossRef Full Text | Google Scholar 18. Fowler JWM, Zhang R, Tao B, Boutagy NE, Sessa WC. Inflammatory stress signaling via NF-kB alters accessible cholesterol to upregulate SREBP2 transcriptional activity in endothelial cells. Elife (2022) 11:e79529. doi: 10.7554/eLife.79529 PubMed Abstract | CrossRef Full Text | Google Scholar 19. He L, Zhang CL, Chen Q, Wang L, Huang Y. Endothelial shear stress signal transduction and atherogenesis: From mechanisms to therapeutics. Pharmacol Ther (2022) 235:108152. doi: 10.1016/j.pharmthera.2022.108152 PubMed Abstract | CrossRef Full Text | Google Scholar 20. Wu YY, Shan SK, Lin X, Xu F, Zhong JY, Wu F, et al. Cellular crosstalk in the vascular wall microenvironment: The role of exosomes in vascular calcification. Front Cardiovasc Med (2022) 9:912358. doi: 10.3389/fcvm.2022.912358 PubMed Abstract | CrossRef Full Text | Google Scholar 21. Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol (2015) 209(1):13–22. doi: 10.1083/jcb.201412052 PubMed Abstract | CrossRef Full Text | Google Scholar 22. Jiang H, Zhou Y, Nabavi SM, Sahebkar A, Little PJ, Xu S, et al. Mechanisms of oxidized LDL-mediated endothelial dysfunction and its consequences for the development of atherosclerosis. Front Cardiovasc Med (2022) 9:925923. doi: 10.3389/fcvm.2022.925923 PubMed Abstract | CrossRef Full Text | Google Scholar 23. Tsiantoulas D, Eslami M, Obermayer G, Clement M, Smeets D, Mayer FJ, et al. APRIL limits atherosclerosis by binding to heparan sulfate proteoglycans. Nature (2021) 597(7874):92–6. doi: 10.1038/s41586-021-03818-3 PubMed Abstract | CrossRef Full Text | Google Scholar 24. Orecchioni M, Kobiyama K, Winkels H, Ghosheh Y, McArdle S, Mikulski Z, et al. Olfactory receptor 2 in vascular macrophages drives atherosclerosis by NLRP3-dependent IL-1 production. Science (2022) 375(6577):214–21. doi: 10.1126/science.abg3067 PubMed Abstract | CrossRef Full Text | Google Scholar 25. Newton K, Dixit VM, Kayagaki N. Dying cells fan the flames of inflammation. Science (2021) 374(6571):1076–80. doi: 10.1126/science.abi5934 PubMed Abstract | CrossRef Full Text | Google Scholar 26. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science (2016) 354(6311):472–7. doi: 10.1126/science.aaf6659 PubMed Abstract | CrossRef Full Text | Google Scholar 27. Gerlach BD, Ampomah PB, Yurdagul A Jr., Liu C, Lauring MC, Wang X, et al. Efferocytosis induces macrophage proliferation to help resolve tissue injury. Cell Metab (2021) 33(12):2445–2463.e8. doi: 10.1016/j.cmet.2021.10.015 PubMed Abstract | CrossRef Full Text | Google Scholar 28. Qiao L, Ma J, Zhang Z, Sui W, Zhai C, Xu D, et al. Deficient chaperone-mediated autophagy promotes inflammation and atherosclerosis. Circ Res (2021) 129(12):1141–57. doi: 10.1161/CIRCRESAHA.121.318908 PubMed Abstract | CrossRef Full Text | Google Scholar 29. Madrigal-Matute J, de Bruijn J, van Kuijk K, Riascos-Bernal DF, Diaz A, Tasset I, et al. Protective role of chaperone-mediated autophagy against atherosclerosis. Proc Natl Acad Sci U.S.A. (2022) 119(14):e2121133119. doi: 10.1073/pnas.2121133119 PubMed Abstract | CrossRef Full Text | Google Scholar 30. Madrigal-Matute J, Cuervo AM, Sluimer JC. Chaperone-mediated autophagy protects against atherosclerosis. Autophagy (2022) p:1–3. doi: 10.1080/15548627.2022.2096397 CrossRef Full Text | Google Scholar 31. Zhang F, Guo X, Xia Y, Mao L. An update on the phenotypic switching of vascular smooth muscle cells in the pathogenesis of atherosclerosis. Cell Mol Life Sci (2021) 79(1):6. doi: 10.1007/s00018-021-04079-z PubMed Abstract | CrossRef Full Text | Google Scholar 32. Wang L, Zhang J, Fu W, Guo D, Jiang J, Wang Y. Association of smooth muscle cell phenotypes with extracellular matrix disorders in thoracic aortic dissection. J Vasc Surg (2012) 56(6):1698–709. doi: 10.1016/j.jvs.2012.05.084 PubMed Abstract | CrossRef Full Text | Google Scholar 33. Zhang DD, Song Y, Kong P, Xu X, Gao YK, Dou YQ, et al. Smooth muscle 22 alpha protein inhibits VSMC foam cell formation by supporting normal LXRα signaling, ameliorating atherosclerosis. Cell Death Dis (2021) 12(11):982. doi: 10.1038/s41419-021-04239-w PubMed Abstract | CrossRef Full Text | Google Scholar 34. Chen J, Kitchen CM, Streb JW, Miano JM. Myocardin: a component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol (2002) 34(10):1345–56. doi: 10.1006/jmcc.2002.2086 PubMed Abstract | CrossRef Full Text | Google Scholar 35. Ostriker AC, Xie Y, Chakraborty R, Sizer AJ, Bai Y, Ding M, et al. TET2 protects against vascular smooth muscle cell apoptosis and intimal thickening in transplant vasculopathy. Circulation (2021) 144(6):455–70. doi: 10.1161/CIRCULATIONAHA.120.050553 PubMed Abstract | CrossRef Full Text | Google Scholar 36. Bell RD, Long X, Lin M, Bergmann JH, Nanda V, Cowan SL, et al. Identification and initial functional characterization of a human vascular cell-enriched long noncoding RNA. Arterioscler Thromb Vasc Biol (2014) 34(6):1249–59. doi: 10.1161/ATVBAHA.114.303240 PubMed Abstract | CrossRef Full Text | Google Scholar 37. Zhao J, Zhang W, Lin M, Wu W, Jiang P, Tou E, et al. MYOSLID is a novel serum response factor-dependent long noncoding RNA that amplifies the vascular smooth muscle differentiation program. Arterioscler Thromb Vasc Biol (2016) 36(10):2088–99. doi: 10.1161/ATVBAHA.116.307879 PubMed Abstract | CrossRef Full Text | Google Scholar 38. Dong K, Shen J, He X, Hu G, Wang L, Osman I, et al. CARMN is an evolutionarily conserved smooth muscle cell-specific LncRNA that maintains contractile phenotype by binding myocardin. Circulation (2021) 144(23):1856–75. doi: 10.1161/CIRCULATIONAHA.121.055949 PubMed Abstract | CrossRef Full Text | Google Scholar 39. Ahmed ASI, Dong K, Liu J, Wen T, Yu L, Xu F, et al. Long noncoding RNA NEAT1 (nuclear paraspeckle assembly transcript 1) is critical for phenotypic switching of vascular smooth muscle cells. Proc Natl Acad Sci U.S.A. (2018) 115(37):E8660–e8667. doi: 10.1073/pnas.1803725115 PubMed Abstract | CrossRef Full Text | Google Scholar 40. Pryma CS, Ortega C, Dubland JA, Francis GA. Pathways of smooth muscle foam cell formation in atherosclerosis. Curr Opin Lipidol (2019) 30(2):117–24. doi: 10.1097/MOL.0000000000000574 PubMed Abstract | CrossRef Full Text | Google Scholar 41. Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M, et al. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res (2014) 115(7):662–7. doi: 10.1161/CIRCRESAHA.115.304634 PubMed Abstract | CrossRef Full Text | Google Scholar 42. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med (2015) 21(6):628–37. doi: 10.1038/nm.3866 PubMed Abstract | CrossRef Full Text | Google Scholar 43. Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol (2019) 16(12):727–44. doi: 10.1038/s41569-019-0227-9 PubMed Abstract | CrossRef Full Text | Google Scholar 44. Sénémaud J, Caligiuri G, Etienne H, Delbosc S, Michel JB, Coscas R. Translational relevance and recent advances of animal models of abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol (2017) 37(3):401–10. doi: 10.1161/ATVBAHA.116.308534 PubMed Abstract | CrossRef Full Text | Google Scholar 45. Kim HW, Shi H, Winkler MA, Lee R, Weintraub NL. Perivascular adipose tissue and vascular Perturbation/Atherosclerosis. Arterioscler Thromb Vasc Biol (2020) 40(11):2569–76. doi: 10.1161/ATVBAHA.120.312470 PubMed Abstract | CrossRef Full Text | Google Scholar 46. Mohanta SK, Peng L, Li Y, Lu S, Sun T, Carnevale L, et al. Neuroimmune cardiovascular interfaces control atherosclerosis. Nature (2022) 605(7908):152–9. doi: 10.1038/s41586-022-04673-6 PubMed Abstract | CrossRef Full Text | Google Scholar 47. Huang Cao ZF, Stoffel E, Cohen P. Role of perivascular adipose tissue in vascular physiology and pathology. Hypertension (2017) 69(5):770–7. doi: 10.1161/HYPERTENSIONAHA.116.08451 PubMed Abstract | CrossRef Full Text | Google Scholar 48. Tuttle T, Darios E, Watts SW, Roccabianca S. Aortic stiffness is lower when PVAT is included: a novel ex vivo mechanics study. Am J Physiol Heart Circ Physiol (2022) 322(6):H1003–h1013. doi: 10.1152/ajpheart.00574.2021 PubMed Abstract | CrossRef Full Text | Google Scholar 49. Tang Y, He Y, Li C, Mu W, Zou Y, Liu C, et al. RPS3A positively regulates the mitochondrial function of human periaortic adipose tissue and is associated with coronary artery diseases. Cell Discovery (2018) 4:52. doi: 10.1038/s41421-018-0041-2 PubMed Abstract | CrossRef Full Text | Google Scholar 50. Mu W, Qian S, Song Y, Yang L, Song S, Yang Q, et al. BMP4-mediated browning of perivascular adipose tissue governs an anti-inflammatory program and prevents atherosclerosis. Redox Biol (2021) 43:101979. doi: 10.1016/j.redox.2021.101979 PubMed Abstract | CrossRef Full Text | Google Scholar 51. Nikolova G, Strilic B, Lammert E. The vascular niche and its basement membrane. Trends Cell Biol (2007) 17(1):19–25. doi: 10.1016/j.tcb.2006.11.005 PubMed Abstract | CrossRef Full Text | Google Scholar 52. Yu D, Liao JK. Emerging views of statin pleiotropy and cholesterol lowering. Cardiovasc Res (2022) 118(2):413–23. doi: 10.1093/cvr/cvab032 PubMed Abstract | CrossRef Full Text | Google Scholar 53. Ling P, Zheng X, Luo S, Ge J, Xu S, Weng J. Targeting angiopoietin-like 3 in atherosclerosis: From bench to bedside. Diabetes Obes Metab (2021) 23(9):2020–34. doi: 10.1111/dom.14450 PubMed Abstract | CrossRef Full Text | Google Scholar 54. Feng X, Zhang L, Xu S, Shen AZ. ATP-citrate lyase (ACLY) in lipid metabolism and atherosclerosis: An updated review. Prog Lipid Res (2020) 77:101006. doi: 10.1016/j.plipres.2019.101006 PubMed Abstract | CrossRef Full Text | Google Scholar 55. Liu Z, Ma X, Ilyas I, Zheng X, Luo S, Little PJ, et al. Impact of sodium glucose cotransporter 2 (SGLT2) inhibitors on atherosclerosis: from pharmacology to pre-clinical and clinical therapeutics. Theranostics (2021) 11(9):4502–15. doi: 10.7150/thno.54498 PubMed Abstract | CrossRef Full Text | Google Scholar 56. Ma X, Liu Z, Ilyas I, Little PJ, Kamato D, Sahebka A, et al. GLP-1 receptor agonists (GLP-1RAs): cardiovascular actions and therapeutic potential. Int J Biol Sci (2021) 17(8):2050–68. doi: 10.7150/ijbs.59965 PubMed Abstract | CrossRef Full Text | Google Scholar 57. Ding Y, Zhou Y, Ling P, Feng X, Luo S, Zheng X, et al. Metformin in cardiovascular diabetology: a focused review of its impact on endothelial function. Theranostics (2021) 11(19):9376–96. doi: 10.7150/thno.64706 PubMed Abstract | CrossRef Full Text | Google Scholar 58. Feng X, Chen W, Ni X, Little PJ, Xu S, Tang L, et al. Metformin, macrophage dysfunction and atherosclerosis. Front Immunol (2021) 12:682853. doi: 10.3389/fimmu.2021.682853 PubMed Abstract | CrossRef Full Text | Google Scholar 59. Deng M, Su D, Xu S, Little PJ, Feng X, Tang L, et al. Metformin and vascular diseases: A focused review on smooth muscle cell function. Front Pharmacol (2020) 11:635. doi: 10.3389/fphar.2020.00635 PubMed Abstract | CrossRef Full Text | Google Scholar 60. Li K, Zemmrich C, Bramlage P, Persson AB, Sacirovic M, Ritter O, et al. Effect of ACEI and ARB treatment on nitric oxide-dependent endothelial function. Vasa (2021) 50(6):413–22. doi: 10.1024/0301-1526/a000971 PubMed Abstract | CrossRef Full Text | Google Scholar 61. Tehrani AY, White Z, Tung LW, Zhao RRY, Milad N, Seidman MA, et al. Pleiotropic activation of endothelial function by angiotensin II receptor blockers is crucial to their protective anti-vascular remodeling effects. Sci Rep (2022) 12(1):9771. doi: 10.1038/s41598-022-13772-3 PubMed Abstract | CrossRef Full Text | Google Scholar 62. Zhang FS, He QZ, Qin CH, Little PJ, Weng JP, Xu SW. Therapeutic potential of colchicine in cardiovascular medicine: a pharmacological review. Acta Pharmacol Sin (2022) p:1–18. doi: 10.1038/s41401-021-00835-w CrossRef Full Text | Google Scholar 63. Dabravolski SA, Sukhorukov VN, Kalmykov VA, Grechko AV, Shakhpazyan NK, Orekhov AN. The role of KLF2 in the regulation of atherosclerosis development and potential use of KLF2-targeted therapy. Biomedicines (2022) 10(2):254. doi: 10.3390/biomedicines10020254 PubMed Abstract | CrossRef Full Text | Google Scholar 64. Fan Y, Lu H, Liang W, Hu W, Zhang J, Chen YE. Krüppel-like factors and vascular wall homeostasis. J Mol Cell Biol (2017) 9(5):352–63. doi: 10.1093/jmcb/mjx037 PubMed Abstract | CrossRef Full Text | Google Scholar 65. Heidary Moghaddam R, Samimi Z, Asgary S, Mohammadi P, Hozeifi S, Hoseinzadeh-Chahkandak F, et al. Natural AMPK activators in cardiovascular disease prevention. Front Pharmacol (2021) 12:738420. doi: 10.3389/fphar.2021.738420 PubMed Abstract | CrossRef Full Text | Google Scholar 66. Engelen SE, Robinson AJB, Zurke YX, Monaco C. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: how to proceed? Nat Rev Cardiol (2022) 19(8):522–42. doi: 10.1038/s41569-021-00668-4 PubMed Abstract | CrossRef Full Text | Google Scholar 67. Man AWC, Li H, Xia N. Impact of lifestyles (Diet and exercise) on vascular health: Oxidative stress and endothelial function. Oxid Med Cell Longev 2020 (2020) p:1496462. doi: 10.1155/2020/1496462 CrossRef Full Text | Google Scholar 68. Bernhard D, Pfister G, Huck CW, Kind M, Salvenmoser W, Bonn GK, et al. Disruption of vascular endothelial homeostasis by tobacco smoke: impact on atherosclerosis. FASEB J (2003) 17(15):2302–4. doi: 10.1096/fj.03-0312fje PubMed Abstract | CrossRef Full Text | Google Scholar 69. Humphrey JD, Schwartz MA. Vascular mechanobiology: Homeostasis, adaptation, and disease. Annu Rev BioMed Eng (2021) 23:1–27. doi: 10.1146/annurev-bioeng-092419-060810 PubMed Abstract | CrossRef Full Text | Google Scholar 70. Ben-Moshe S, Veg T, Manco R, Dan S, Papinutti D, Lifshitz A, et al. The spatiotemporal program of zonal liver regeneration following acute injury. Cell Stem Cell (2022) 29(6):973–989.e10. doi: 10.1016/j.stem.2022.04.008 PubMed Abstract | CrossRef Full Text | Google Scholar 71. Manco R, Itzkovitz S. Liver zonation. J Hepatol (2021) 74(2):466–8. doi: 10.1016/j.jhep.2020.09.003 PubMed Abstract | CrossRef Full Text | Google Scholar Keywords: aorta zonation, atherosclerosis, homeostasis, vascular ecosystem, endothelial function Citation: Xu S, Lyu QR, Ilyas I, Tian X-Y and Weng J (2022) Vascular homeostasis in atherosclerosis: A holistic overview. Front. Immunol. 13:976722. doi: 10.3389/fimmu.2022.976722 Received: 23 June 2022; Accepted: 18 August 2022;Published: 12 September 2022. Edited by: Xiaofeng Yang, Temple University, United StatesReviewed by: Jian Xu, University of Oklahoma Health Sciences Center, United StatesYifan Lu, Temple University, United StatesCopyright © 2022 Xu, Lyu, Ilyas, Tian and Weng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms. *Correspondence: Suowen Xu, [email protected]; Jianping Weng, [email protected] †These authors have contributed equally to this work ‡ORCID: Suowen Xu, orcid.org/0000-0002-5488-5217, Jianping Weng orcid.org/0000-0002-7889-1697, Xiao-Yu Tian orcid.org/0000-0003-3472-9898 This article is part of the Research TopicEndothelial Cells as Innate Immune Cells View all 7 Articles |
【本文地址】